Two major types: Autosomal Dominant Polycystic Kidney Disease (Adult
Polycystic Kidney Disease [APKD] and Autosomal Recessive Polycystic
Kidney Disease (Infantile Polycystic Kidney Disease)
Autosomal Dominant
Polycystic Kidney Disease
-
Is a slowly progressive disease with
nearly 100% penetrance
-
Potter Type III
-
Cause: gene located on short arm of
chromosome 16 (in 90%
-
Incidence:1:1,000 people carry the
mutant gene
-
Histo: abnormal rate of tubule
divisions (Potter Type III) with hypoplasia of portions of tubules
left behind as the ureteral bud advances; cystic dilatation of Bowman
capsule, loop of Henle, proximal convoluted tubule, coexisting with
normal tissue
-
Mean age at diagnosis: 43 years
(neonatal / infantile onset has been reported)
-
M:F = 1:1
-
Onset of cyst formation:
-
54% in 1st decade
-
72% in 2nd decade
-
86% in 3rd decade
Associated with:
· Cysts in: liver (25-50%), pancreas (9%)
· Aneurysm: saccular "berry" aneurysm of
cerebral arteries (3-13%)
· Mitral valve prolapse
· Hypertension (50-70%)
· Azotemia
· Hematuria, proteinuria
· Lumbar / abdominal pain
· Bilaterally large kidneys with multifocal
round lesions; unilateral enlargement may be the first manifestation of
the disease
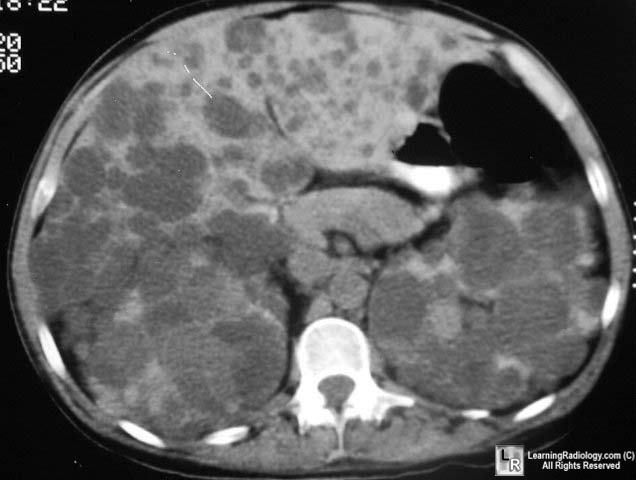
Adult Polycystic Kidney Disease Multiple low attenuation cystic lesions are seen in both
kidneys and throughout the liver.
· Cysts may calcify in curvilinear rim- /
ringlike irregular amorphous fashion elongated + distorted + attenuated
collecting system nodular puddling of contrast material on delayed
images
· "Swiss cheese" nephrogram = multiple lesions
of varying size with smooth margins
· Polycystic kidneys shrink after beginning of
renal failure, after renal transplantation, or on chronic hemodialysis
· NUC: poor renal function on Tc-99m DTPA scan
US
· Multiple cysts in cortical region (usually
not seen prior to teens)
· Diffusely echogenic when cysts small
(children)
· Renal contour poorly demarcated
OB-US
· Large echogenic kidneys similar to infantile
PCKD (usually in 3rd trimester, earliest sonographic diagnosis at 14
weeks), can be unilateral
· Macroscopic cysts (rare)
· Normal amount of amniotic fluid /
oligohydramnios (renal function usually not impaired)
Complications
· Death from uremia (59%) / cerebral
hemorrhage (secondary to hypertension or ruptured aneurysm [13%]
· Renal calculi
· Urinary tract infection
· Cyst rupture
· Hemorrhage
· Renal cell carcinoma (increased risk)
Differential
Diagnosis
· Multiple simple cysts (less diffuse, no
family history)
· von Hippel-Lindau disease (cerebellar
hemangioblastoma, retinal hemangiomas, occasionally pheochromocytomas)
· Acquired uremic cystic disease (kidneys
small, no renal function, transplant)
· Infantile PCKD (usually microscopic cysts)
Autosomal Recessive
Polycystic Kidney Disease = Polycystic Disease of Childhood
· Potter Type I
· Incidence:1: 6,000 to 1:50,000 livebirths
· F > M; carrier frequency of 1:112
Pathology
· Kidney: abnormal proliferation + dilatation
of collecting tubules resulting in multiple 1- to 2-mm cysts
· Liver: periportal fibrosis often with
abnormal proliferation + dilatation of bile ducts
· Pancreas: pancreatic fibrosis
ANTENATAL FORM (most
common)
· 90% of tubules show cystic changes
· Onset of renal failure in utero
· Potter sequence
· Oligohydramnios and dystocia (large
abdominal mass)
· Prognosis: death from renal failure /
respiratory insufficiency (pulmonary hypoplasia) within 24 hours in 75%,
within 1 year in 93%; uniformly fatal
NEONATAL FORM
· 60% of tubules show ectasia + minimal
hepatic fibrosis + bile duct proliferation
· Onset of renal failure within 1st month of
life
· Prognosis: death from renal failure /
hypertension / left ventricular failure within 1st year of life
INFANTILE FORM
· 20% of renal tubules involved + mild /
moderate periportal fibrosis
· Disease appears by 3-6 months of age
· Prognosis: death from chronic renal failure
/ systemic arterial hypertension / portal hypertension
JUVENILE FORM
· 10% of tubules involved + gross hepatic
fibrosis + bile duct proliferation
· Disease appears at 1-5 years of age
· Prognosis: death from portal hypertension
· The less severe the renal findings, the more
severe the hepatic findings!
· Severe pulmonary hypoplasia
· Pneumothorax / pneumomediastinum
Liver
· Portal venous hypertension
· Tubular cystic dilatation of small
intrahepatic bile ducts
· Increase in liver echogenicity (from
congenital hepatic fibrosis)
Kidneys
· Bilateral gross renal enlargement
· Faint nephrogram + blotchy opacification on
initial images
· Increasingly dense nephrogram
· Poor visualization of collecting system
· "Sunburst nephrogram" = striated nephrogram
with persistent radiating opaque streaks (collecting ducts) on
· Delayed images
· Prominent fetal lobulation
CT
· Prolonged corticomedullary phase
US
· Hyperechoic enlarged kidneys (unresolved 1-
to 2-mm cystic / ectatic dilatation of renal tubules increase number of
acoustic interfaces)
· Increased renal through-transmission (high
fluid content of cysts)
· Loss of corticomedullary differentiation,
poor visualization of renal sinus + renal borders
· Occasionally discrete macroscopic cysts <1
cm
· Compressed / minimally dilated collecting
system
OB-US (diagnostic as early as 17 weeks GA):
· Progressive renal enlargement with renal
circumference : abdominal circumference ratio >0.30
· Hyperechoic renal parenchyma
· Nonvisualization of urine in fetal bladder
(in severe cases)
· Oligohydramnios (33%)
· Small fetal thorax
OB management
· Chromosome studies to determine if other
malformations present (e.g., trisomy 13 / 18)
· Option of pregnancy termination <24 weeks
· Nonintervention for fetal distress >24 weeks
if severe oligohydramnios present
· Risk of recurrence:25%
· DDx: Meckel-Gruber syndrome, adult
polycystic kidney disease
